bCNN-Methylpred:基于特征的基于分支卷积神经网络的RNA序列修饰预测
-
- 摘要
- 1、介绍
- 2、相关研究
-
- (1)CNN的一个新分支:该网络结合了不同编码方案的特征,准确地预测了不同RNA序列中的m6a位点
- (2)一种新的编码方案:我们提出了一种新的环状编码方案,它考虑了RNA序列中四个核苷酸碱基的每一个可能的组合,即腺嘌呤(A)、胞嘧啶(C)、鸟嘌呤(G)和尿嘧啶(U)。该编码方案进一步提高了对不同RNA序列中m6a位点的预测准确性。
- (3)特征融合:该方法采用三种编码方案,使用单个编码的RNA序列,然后结合它们的特征。随后,利用这些组合特征来识别不同RNA序列中的m6a位点。
- (4)对所提出的模型的生物学解释:我们从生物学的角度来研究所提出的模型,通过解释基于一个完全成熟的解释程序的训练模型,即在硅突变中。
- 3、材料和方法
-
- 3.1 编码方案
-
- 3.1.1.循环编码的一种算法
- 3.1.2.循环编码的示例
- 3.2.建议的方法
- 3.3.基准数据集
-
- 3.3.1.四个基准数据集
- 3.3.2.miCLIP-Seq数据集
- 3.4.性能评估
-
- 3.4.1对四个基准数据集的预测
- 比较m6a地点预测使用提出的和最先进的方法基于智人物种。
- 比较m6a地点预测使用提出的和最先进的方法基于肌肉杆菌物种。
- .比较m6a地点预测使用提出的和最先进的方法基于酿酒酵母物种。
- 利用基于拟南芥物种的现有方法和现有方法进行m6a位点预测的比较。
- 3.4.3.对miCLIP-Seq数据集的预测
-
- .比较m6a使用基于cnn的方法和基于人类和小鼠物种的最先进的机器学习方法进行站点预测。
摘要
RNA修饰在各种细胞和生物过程中都至关重要。在现有的RNA修饰中,n6-甲基腺苷(m6a)被认为是最重要的修饰,因为它参与了许多生物过程的修饰。m6a位点的预测是至关重要的,因为它可以更好地理解其功能机制。在这方面,虽然实验方法是有用的,但它们却很耗时。以前,研究人员曾试图使用计算方法来预测m6a位点,以克服实验方法的局限性。其中一些方法是基于经典的机器学习技术,它们依赖于手工制作的特性,并需要领域知识,而其他的方法则是基于深度学习的。然而,这两种方法都缺乏鲁棒性,产生的精度较低。因此,我们开发了一个基于分支的卷积神经网络和一个新的RNA序列表示。该网络会自动从指定输入的每个分支中提取特征。随后,将这些特征连接到特征空间中,以预测m6a站点。最后,我们用四种不同的物种进行了实验。该方法优于现有的现有方法,对智人、 H. sapiens, M. musculus,
S. cerevisiae, and A. thaliana datasets的准确率分别为94.91%、94.28%、88.46%和94.8%
1、介绍
基因表达是指利用基因信息合成功能基因产物。这是一个多层的过程,首先控制DNA中编码的特定序列的信息,然后复制到RNA分子中。随后,RNA分子发生分支,将其序列信息转移到多肽(编码RNA、mrna)或非编码RNA中。更重要的是,RNA的功能不仅依赖于序列信息,还依赖于剪接;特别是,选择性剪接可以使RNA多样化。每个RNA核苷酸都可以进行化学修饰或交换(RNA编辑)。自1957年发现第一种RNA修饰以来,已有超过150种RNA修饰被记录下来。在这些修饰阳离子中,N6甲基腺苷(m6a)是存在于各种物种中最丰富和最典型的修饰物。它被认为与多种生物过程密切相关,包括RNA定位和降解[1]、RNA结构动力学[2]、选择性剪接[3]、初级microRNA加工[4]、细胞分化和重编程[5],以及昼夜节律的调控。例如,m6a是关联包括癌症、肥胖型[7]、急性髓细胞白血病[8]、寨卡病毒[9]和抑郁症[10]。除m6a外,m1a还与x-连锁的顽固性癫痫、多性呼吸链缺陷[11]和神经发育退化[12]相关。A-to-i与癌症[13,14]和神经系统疾病[15]相关。m5C与乳腺癌[16]和智力残疾综合征[17,18]相关。关于RNA修饰和疾病之间关系的进一步细节可在[19,20]中获得。
2、相关研究
考虑到RNA修饰的重要性,特别是m6a,位点识别需要更好地理解其功能机制。高通量实验技术,如MERIP和m6A-seq,已分别在[21,22]中提出。MERIP[21]鉴定出含有m6a的哺乳动物基因的mRNA,表明m6a是一种典型的mRNA碱基修饰。研究发现,m6a位点在终止密码子附近和三个主要的非翻译区富集。基于大规模平行测序和抗体介导的捕获,m6a-seq[22]以转录组范围的方式提供了小鼠和人类的m6a修饰景观。据报道,m6a位点出现在终止密码子周围和长内外显子内。然而,这些方法存在以下局限性:(1)定位m6a位点正确位置的精度低,(2)计算复杂度高,(3)限制了m6a位点大规模识别的适用性。因此,需要准确、快速的方法来正确识别m6a位点[23]。
最近,基于深度学习(DL)和机器学习(ML)的修改预测方法已经被开发出来来识别m6a位点。[24]报道了一项使用ML方法的开创性研究,其中提出了“irna-甲基”用于m6a位点鉴定。在这种方法中,作者利用了“伪二核苷酸组成”,在构建RNA序列时,其中结合了三种RNA的物理化学性质。为了开发他们的m6a站点预测器模型,他们使用了一个支持向量机。在[25]中,作者提出了pRNAm-PC,其中RNA序列样本通过加入一种额外的伪二核苷酸组成模式来表达,其成分通过一系列自协方差和交叉协方差转换从物理-化学矩阵中衍生而来。在[26]中,作者提出了一种名为rna-甲基pred的生物信息学模型。他们通过结合双轮廓贝叶斯、双核苷酸组成和三个特征提取的k-最近邻分数来开发他们的模型,与以前只使用单一特征描述符的方法相比,产生了改进的结果。在[27]中,作者表明,使用二进制编码方案和k-mer频率的组合可以提高性能。在[28]中,作者提出了一个名为“SRAMP”的强大的预测工具。在他们提出的方法中,使用了多种类型的特征描述符,包括核苷酸序列的位置二进制编码、k-最近邻编码、核苷酸对谱编码和二级结构模式来训练基于随机森林的集成预测模型,用于m6a位点识别。与其他现有的预测器相比,他们提出的方法取得了相对更好的性能。在[29]中提出了一种名为“rnamethypre”的新的m6A位点预测因子,其中成分信息和位置特异性信息被用于开发预测人类和小鼠物种中m6A位点的预测模型。在[30]中提出了一种基于dl的潜在特征生成算法,以提高预测性能。在[31]中,作者提出了一种基于序列的预测器,用于检测多个物种RNA序列中的m6a位点。他们提出了一种基于编码编码算法的特征表示算法。他们将F-score算法与序列前向搜索相结合,以优化特征空间,提高表示能力。他们应用XGBoost算法,使用可用的最优特征进行模型训练。最近,纳扎里等人。提出了一种基于DL的卷积神经网络(CNN),他们将其命名为iN6-甲基(五步)[35],用于对智人、肌肉分枝杆菌和酿酒酵母的基准物种的m6a位点预测在他们提出的方法中,他们使用一个基于自然语言处理的word2vec模型来提取特征。在这种方法中,使用k-mer技术将每个序列手工分割成长度为k的单词。他们将k的值设置为3,每个单词都被映射到它相应的特征表示中。由于他们的模型使用整个基因组进行训练,它的计算复杂度很高,而它对m6a位点的预测却很慢。最近,[36]的作者提出了一种基于cnn的架构来识别RNA序列中的m6a位点,他们将其命名为pm6A-CNN。他们使用了单热编码和核苷酸化学性质(NCP)的组合作为模型的输入。此外,他们还使用了一种网格搜索算法来确定其模型的最优参数。与现有的方法相比,他们提出的方法取得了更好的性能。然而,如果特征表示是高度非线性的,则将特征作为模型输入的组合在训练方面存在局限性。
本文的主要贡献如下:
(1)CNN的一个新分支:该网络结合了不同编码方案的特征,准确地预测了不同RNA序列中的m6a位点
(2)一种新的编码方案:我们提出了一种新的环状编码方案,它考虑了RNA序列中四个核苷酸碱基的每一个可能的组合,即腺嘌呤(A)、胞嘧啶?、鸟嘌呤(G)和尿嘧啶(U)。该编码方案进一步提高了对不同RNA序列中m6a位点的预测准确性。
(3)特征融合:该方法采用三种编码方案,使用单个编码的RNA序列,然后结合它们的特征。随后,利用这些组合特征来识别不同RNA序列中的m6a位点。
(4)对所提出的模型的生物学解释:我们从生物学的角度来研究所提出的模型,通过解释基于一个完全成熟的解释程序的训练模型,即在硅突变中。
3、材料和方法
本节介绍了不同的编码方案、所提出的方案、基准数据集、性能评估、讨论和模型的生物学解释。
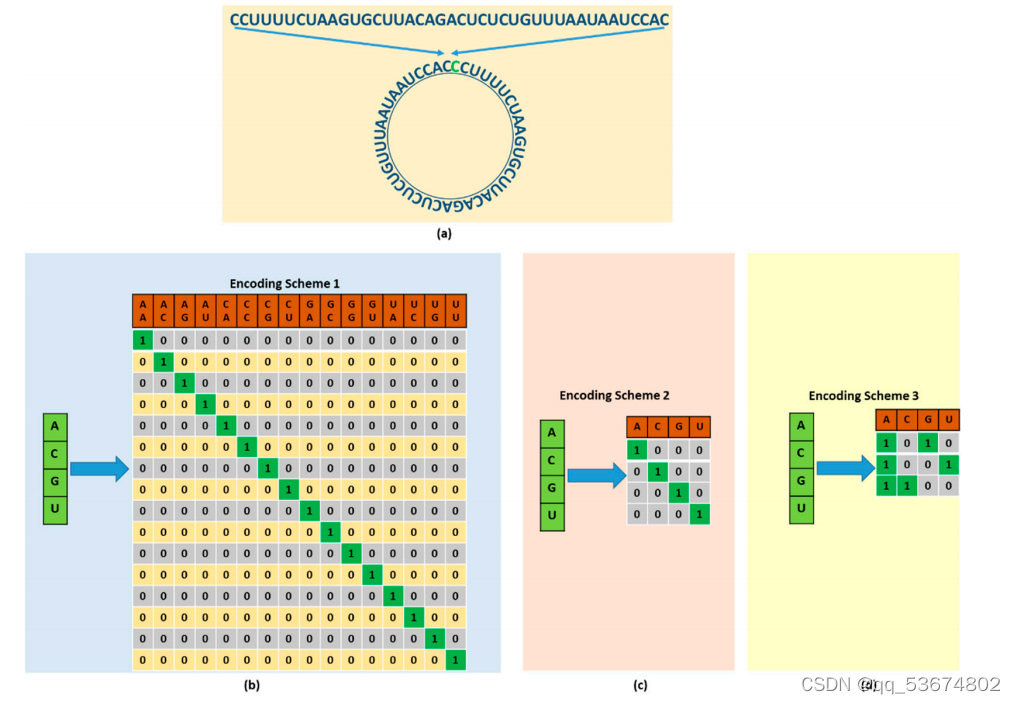
3.1 编码方案
以深度神经网络所能接受的形式表示RNA序列是最基本和最重要的步骤。因此,我们使用了一种新的循环编码方案来表示RNA序列,并使用了两种典型的编码技术来表示RNA序列。编码方案如图1所示。所提出的环状编码方案是基于RNA序列中四个核苷酸碱基的成对组合。首先,我们将序列转换为一个圆形的形状,如图1a所示;随后,我们将其编码为一个成对的组合。我们有一个特定长度的核苷酸碱基a、C、G和U的序列。为了制作配对的核苷酸碱基,我们从左边开始挑选碱基并制作成对。为了使这个组合变成循环,我们复制了序列末端的第一个核苷酸碱基,并使修改后的序列成对。一旦这些对形成,我们就为每一个对分配唯一的代码。该编码方案考虑了所有可能的核苷酸碱基对组合,从而得到16种可能的构型。每对对应的位被设置为1,而其余的位被设置为零。循环编码的总体配置如图1b所示。由于循环编码考虑了RNA序列中核苷酸碱基的每一种可能的配置和排列,因此可以利用更多的信息更有效地训练该模型。 因此,该模型将在预测RNA序列中的修饰位点方面产生更准确的结果。下面的性能评估部分将讨论和全面评估包含循环编码的优点单热编码是一种二进制表示法四种核苷酸碱基A、C、G、U,其中核苷酸碱基A、C、G、U分别表示为(1,0,0,0)、(0,1,0,0)、(0,0,1,0)、(0,0,0,1)。图1c显示了对单热编码的详细描述。NCP是基于三维笛卡尔坐标系中的三个化学基团的RNA序列中每个核苷酸碱基的表示。RNA序列中的四个核苷酸碱基A、C、G和U都具有不同的化学性质。考虑到环的结构,A和G是由两个环组成的嘌呤在f二级结构中,A和U之间的氢键较弱,而C和G之间的氢键较强。同样,在化学功能方面,A和C属于氨基,而G和U属于酮基。考虑到这三种化学性质,RNA序列可以将笛卡尔坐标系中的四个核苷酸碱基分为三个不同的组。如果?-、?-和?-坐标表示环状结构、氢键和化学函数分别表示环状结构、氢键和化学功能,则每个核苷酸碱基可以由(xi、yi、zi)编码,如下图所示
3.1.1.循环编码的一种算法
1.选择一个序列;2。复制序列末端的第一个核苷酸碱基;3。在给定的序列中形成配对的核苷酸碱基。为每对代码分配唯一的二进制代码(代码分配如图1b所示)。生成的代码被用作网络的输入。
3.1.2.循环编码的示例
1.给定一个序列为:CCUUUUCUAAGUGCUUACAGACUCUCUGUUUAAUAAUCCAU;2。在末端复制第一个基。修改后的序列为:CCUUUUCUAAGUGCUUACAGACUCUCUGUUUAAUAAUCCAUC;4。制造对碱基:(CC)、(UU)、(UU)、(CU)、(AA)、(GU)、(GC)、(UU)、(AC)、(AG)、(AC)、(UC)、(UC)、(UG)、(UU)、(UU)(UA)、(AU)、(AA)、(UC)、(CA)、(UC);5。为每对代码分配二进制代码,如图1b所示。
3.2.建议的方法
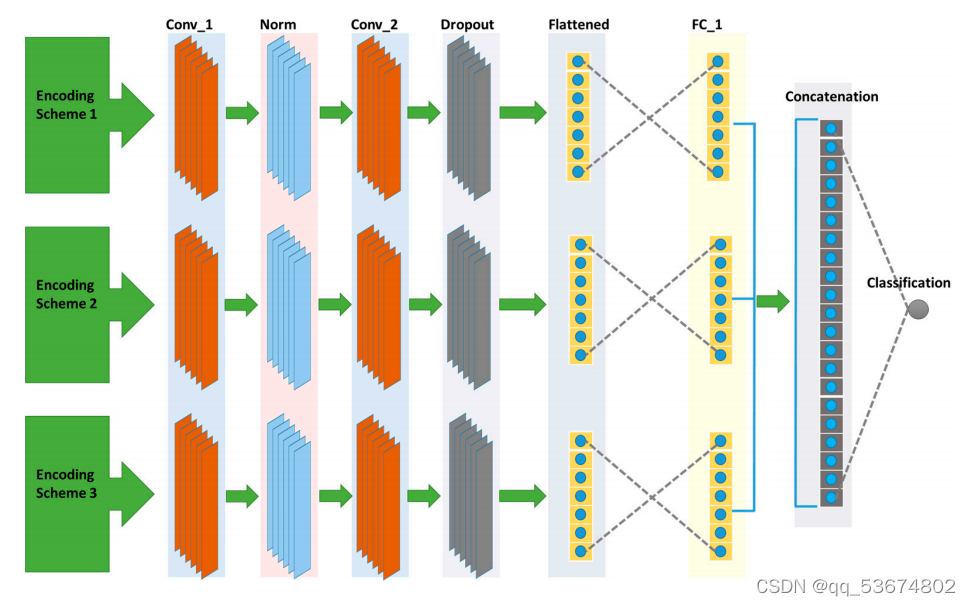
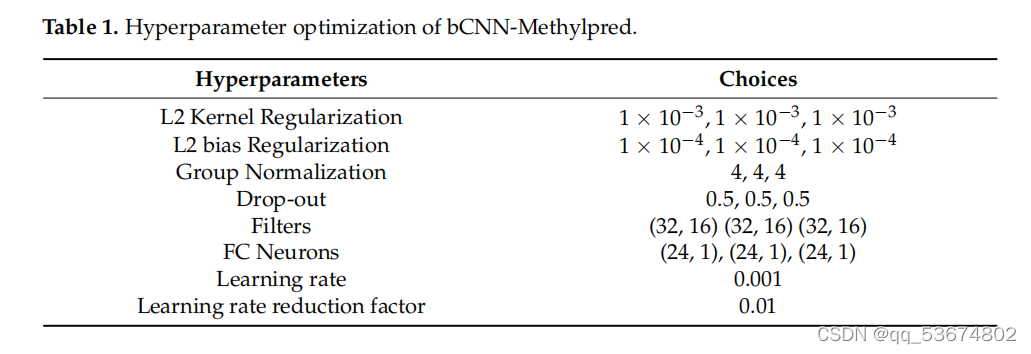
作为解决上述问题的方法,我们提出了bcnn-甲基制备。该网络由三个投影分支组成,如图2所示。第一个分支使用循环编码作为输入,并将它们投影到相应的特性中。第二个分支使用一个热编码作为输入,并在特征空间中生成特征。类似地,第三个分支使用NCP作为输入,并将它们投射到特性空间中所需的特性中。然后,将该网络的所有分支所产生的特征连接到一个特征空间中,然后在最后一个全连接层中分为正序列和负序列。阳性序列和阴性序列分别显示ma6修饰位点的存在和缺失。与非线性输入空间和不可分的输入空间相比,特征空间是线性可分的,因此连接特征而不是直接输入空间是线性可分的。为了进一步说明这一点,RNA序列首先使用三种编码方案进行编码,包括单热编码、循环编码和核苷酸化学性质编码。一旦编码完成,下一步就是在网络中使用这个编码的序列并预测修饰位点。之前的方法首先将编码的序列合并,然后将组合后的序列作为网络的输入,以预测修饰位点。这种组合是线性的,即编码序列不通过非线性函数,而在我们的方法中,我们首先通过单个编码序列通过神经网络。请注意,具有多层和激活函数的神经网络作为非线性函数近似。然后将每个分支的非线性输出组合起来,作为最后一个全连接层的输入,用于预测给定RNA序列中的修饰位点。考虑到所提出的网络配置,每个分支包含两个卷积层、一组归一化层、一个辍学层、一个全连接层和一个输出分类层。bcnn-甲基基的第一、第二和第三分支的输入分别是一个16位向量、4位向量和3位向量。所述网络中每个分支的第一卷积层包括32个滤波器,每个滤波器的大小为5。第一个卷积层的输出通过ReLU激活函数传递。激活函数的输出通过组大小为4的组规范化层[37]传递。第二个卷积层使用组归一化的输出作为输入。这个卷积层有16个带有滤波器的滤波器尺寸为3号。第二卷积层的输出通过ReLU激活函数,然后将该函数平坦为一维特征向量。随后,将一维特征向量通过一个辍学率为0.5的辍学层。辍学层的输出通过具有24个特征的第一个全连接层。每个分支的第一个完全连接的层被连接起来形成72个隐藏的特征。随后,这些隐藏的单元被连接到分类层上。分类层有一个输出神经元来执行二元分类,以确定RNA序列是否发生了修饰换句话说,它表示RNA序列中m6a位点的存在或缺失。我们使用了一个非线性的s型激活函数来进行分类层的输出。此外,我们使用l2正则化来避免网络的过拟合。为了训练网络的参数,我们使用了学习率为0.001的Adam优化器。我们将批处理大小设置为32,并使用基于验证损失的早期停止来进行最大的训练迭代次数,即1000次。bcnn-甲基预网络使用Keras,一个开源DL库,在一个基于gpu的PC上实现,包括一个英特尔?核心i9-9940XCPU,132.0GB内存和四个NVIDIAGeForceRTX2080Ti显卡。bCNN甲基预备网络是轻量级的,可以在任何平台上运行,而无需使用GPU。此外,它还需要极小的内存容量,为284.2kB。因此,该网络不受计算复杂度和内存的影响。虽然我们使用了四个NVIDIAGeForceRTX2080Ti显卡,但唯一的目的是同时操作多个网络,使用不同的物种进行训练。换句话说,它表示RNA序列中m6a位点的存在或缺失。我们使用了一个非线性的s型激活函数来进行分类层的输出。此外,我们使用l2正则化来避免网络的过拟合。为了训练网络的参数,我们使用了学习率为0.001的Adam优化器。我们将批处理大小设置为32,并使用基于验证损失的早期停止来进行最大的训练迭代次数,即1000次。bcnn-甲基预网络使用Keras,一个开源DL库,在一个基于gpu的PC上实现,包括一个英特尔?核心i9-9940XCPU,132.0GB内存和四个NVIDIAGeForceRTX2080Ti显卡。bCNN甲基预备网络是轻量级的,可以在任何平台上运行,而无需使用GPU。此外,它还需要极小的内存容量,为284.2kB。因此,该网络不受计算复杂度和内存的影响。虽然我们使用了四个NVIDIAGeForceRTX2080Ti显卡,但唯一的目的是同时操作多个网络,使用不同的物种进行训练关于超参数优化的更多细节见表1
3.3.基准数据集
为了通过预测RNA序列中的m6a位点来分析所提网络的性能,我们首先选择了4个不同的物种基准数据集,然后是miCLIP-Seq基准数据集。
3.3.1.四个基准数据集
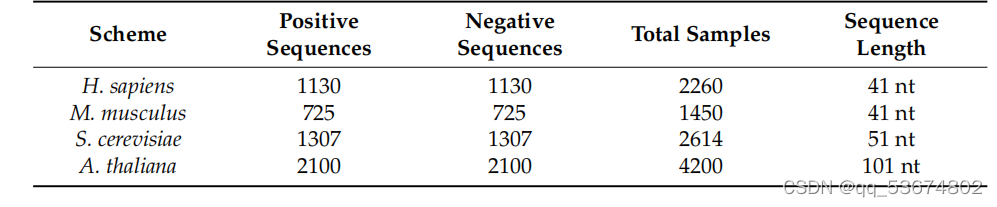
本研究中使用的数据集包括智、人、肌肉分枝杆菌、酿酒酵母和拟南芥的数据集。这四个基准数据集的所有序列都在中心包含A。阳性序列以真实的m6a位点为中心,而阴性序列不含m6a位点。智人基准数据集是由[38]的作者生成的。该数据集由1130个阳性序列和1130个阴性序列组成,其中每个序列的长度为41nt。在[22]中,作者准备了一个肌肉杆菌基准数据集。该数据集中每个序列的长度也为41nt。肌肉杆菌基准数据集包括725个阳性序列和725个阴性序列。酿酒酵母基准数据集是由[39]的作者开发的。该数据集包含1307个阳性序列和1307个阴性序列。每个序列的长度为51nt。拟南芥的基准数据集是由[40]的作者创建的。它包含2100个阳性序列和2100个阴性序列,其中每个序列的长度为101nt。表2给出了这些基准数据集的摘要。我们使用k倍交叉验证来评估所提方法的性能。根据最近的文献调查,使用k倍交叉验证或折刀检验对模型的评估不需要专门的测试数据集。不同的k倍结果可以被认为是不同的独立测试数据集。
3.3.2.miCLIP-Seq数据集
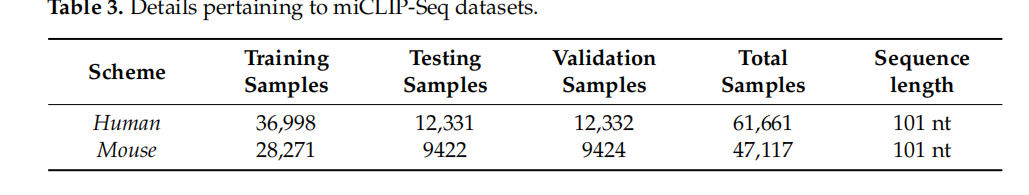
接下来,我们选择了可以在单碱基分辨率下识别m6a位点的miCLIP-Seq数据集。我们从与人类和小鼠[41,42]的SRAMP相同的来源获得了该数据集,其中包括五种细胞系和组织类型,即A549、CD8T、HEK293、大脑和肝脏。为了生成阳性和阴性样本,我们执行了[43]中详细说明的程序。对于阳性样本,我们定义了101nt的包含m6a位点的序列。我们首先利用集成数据库(http://www.ensembl.org/,2021年7月27日访问)将m6A位点定位到该基因的最长的转录本,然后在固定大小的窗口中随机定位m6A位点。随后,我们提取了长度为101nt的周围序列。由于有报道称m6A位点会聚集[21],为了在随机定位m6A位点之前避免样本冗余,我们首先将它们在50nt内合并,并在合并后的位点中选择居中的一个。为了生成阴性样本,我们选择了不包含m6a位点的窗口附近的长度。这些窗口通过10nt和100步生成,得到最接近阳性样本的相应阴性样本。对于阳性样本两侧最接近的两个样本,我们随机选择一个样本。对这些数据集的进一步描述如表3所示。
3.4.性能评估
为了评估该方法在RNA序列中m6a位点方面的有效性,我们使用上述6个不同的基准数据集进行了广泛的比较定量分析。为了进行性能评估,首先使用第3.3.1节和第3.3.2节中讨论的方法生成这些序列。一旦生成了正序列和负序列,我们将它们以模型可接受的格式进行编码,即使用上面讨论的三种编码方案(即循环编码、单热编码和NCP编码)的二进制格式。随后,它们被用作拟议网络的输入。该网络由三个分支组成,其中第一个分支使用循环编码编码的序列,第二个分支使用单热编码方案编码的序列,最后一个分支使用NCP编码的序列。对这3种RNA序列编码方案的详细描述见第3.1节。在对RNA序列进行编码后,我们独立训练了个体物种上所提出的网络,并预测了特定物种中的修饰位点
3.4.1对四个基准数据集的预测
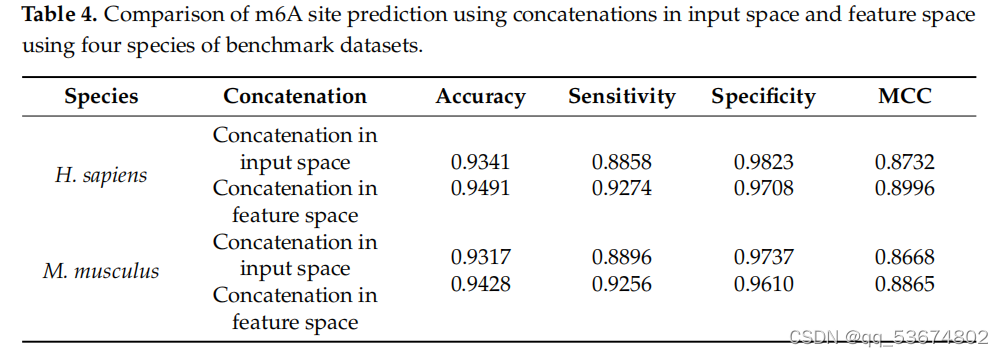
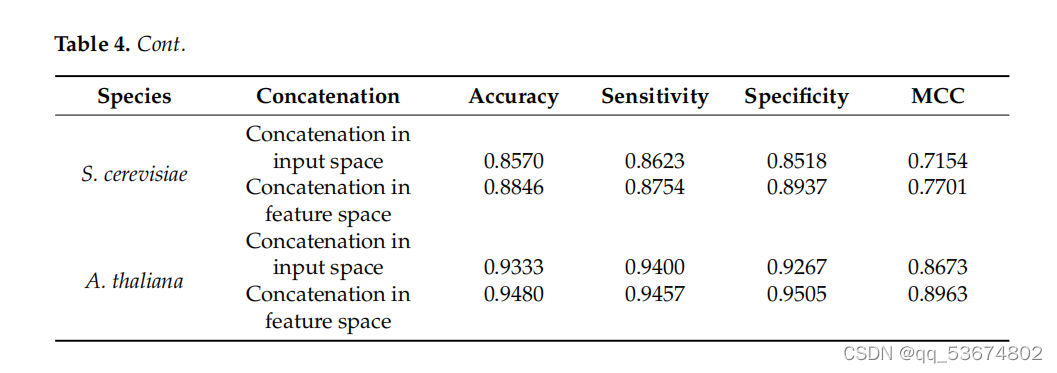
通过不同的实验来评估该网络的有效性。在第一个实验中,我们通过直接连接三种编码方案的输入表示来分析所提出的网络;随后,我们将连接的输入输入到一个分支CNN来预测m6a站点。在这一步中,我们首先连接所有三个编码,然后将连接后的编码作为网络的输入。随后,我们使用提出的三分支CNN,在RNA分支中分别使用三个输入表示,预测RNA序列中的m6a位点。在三个分支的CNN中,输入表示没有连接到输入中,而是将每个输入的特征连接到网络的特征空间中。值得注意的是,CNN将非线性输入空间转换为线性特征空间。因此,我们将线性特征连接到相应分支的特征空间中,并使用连接后的特征来检测m6a站点。T中列出了所有四种物种在输入空间和特征空间中的串联所产生的结果。根据我们的分析,我们观察到三分支CNN比单分支CNN预测更准确。这验证了连接特性而不是直接输入会产生更健壮和准确的模型。除了在输入空间和特征空间中执行连接外,我们还评估了所有三种特征在不同组合中的有效性。在第一个组合中,我们结合了由单热编码和NCP生成的特征。随后,我们根据不同的评价指标来评估模型的性能,如表5所示。在第二个组合中,我们使用了带有循环编码的单热编码。最后,在第三个组合中,我们使用了循环编码和NCP组合。我们观察到,循环编码与单热和NCP的结合表明,在准确性、灵敏度、特异性、MCC和AUC方面略有改善,如表5所示。随后,我们评估了个体编码方案对肌肉分枝杆菌物种RNA序列中m6a位点预测的影响,如表6所示。之前使用pm6A-CNN[36]评估了单热和NCP编码方案对RNA序列修饰预测的影响。然而,为了证明环状编码的效果,我们对个体编码方案进行了比较定量分析,以预测RNA序列修饰。我们观察到,与[36]中所列的单热编码和NCP相比,循环编码提高了准确性、灵敏度和MCC
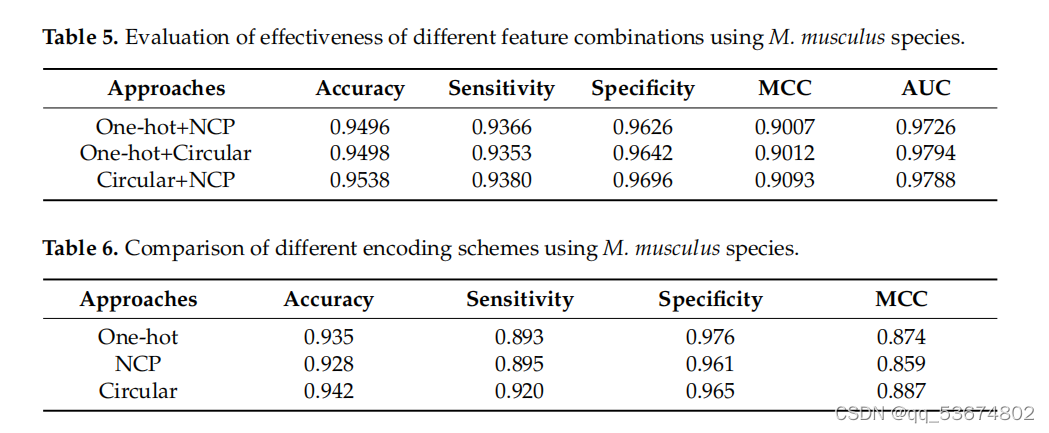
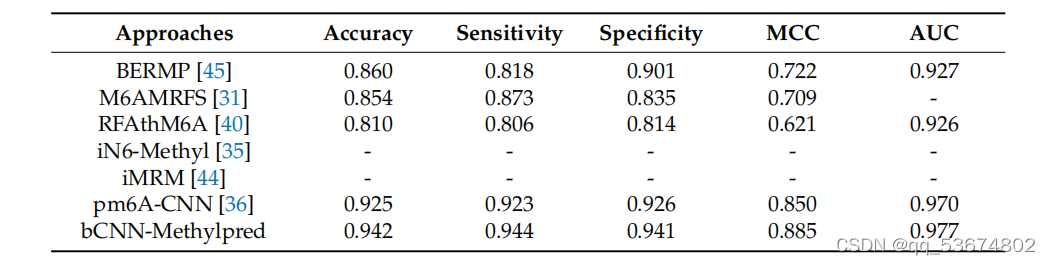
接下来,我们利用智人和肌肉物种,对现有的最先进的方法如M6AMRFS[31]、iN6-Metel[35]、iMRM[44]和pm6A-CNN[36]进行了比较定量分析。分析结果见表7和表8。考虑到智人物种的定量分析(如表7所示),我们观察到该方法在准确性、灵敏度和MCC方面分别比M6AMRFS高出3.12%、10.92%和5.02%。该方法的准确性、灵敏度、MCC和AUC分别比iMRM高3.02%、14.12%、4.82%和7.6%,分别比iMRM高3.12%、10.52%、6.32%和3.9%。同样,该方法的准确率、灵敏度、MCC和AUC分别比pm6A-CNN分别高出0.52%、4.32%、0.52%和1.9%。同样,考虑到肌肉分枝杆菌的种类,所提出的方法在准确性、灵敏度和AUC方面表明,与表8中列出的所有竞争对手相比,性能都有所提高。其对两种方法的特异性均高于其他方法,.我们观察到,其他方法的特异性高于所提出的方法,但敏感度明显较低。这并不是一个衡量模型整体准确性的好指标。相比之下,该方法具有较高的灵敏度和特异性,且两者之间的差异不显著,因此,总体准确率较高。
比较m6a地点预测使用提出的和最先进的方法基于智人物种。
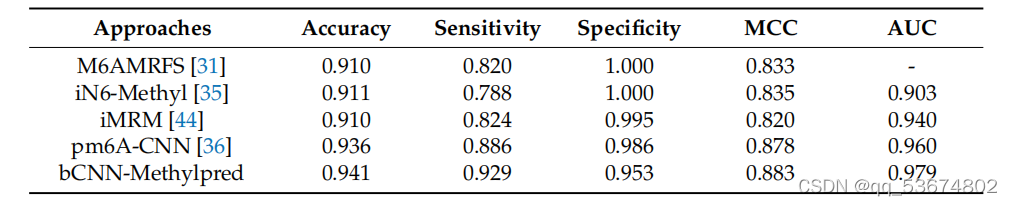
比较m6a地点预测使用提出的和最先进的方法基于肌肉杆菌物种。
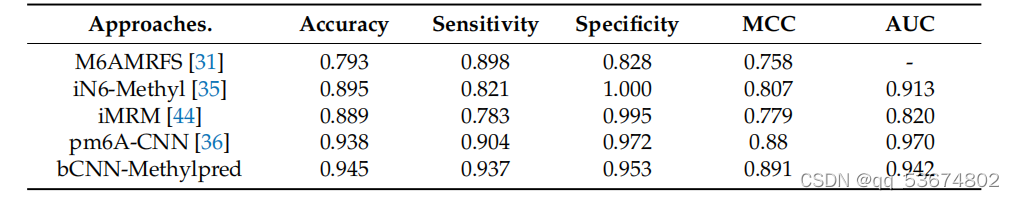
.比较m6a地点预测使用提出的和最先进的方法基于酿酒酵母物种。
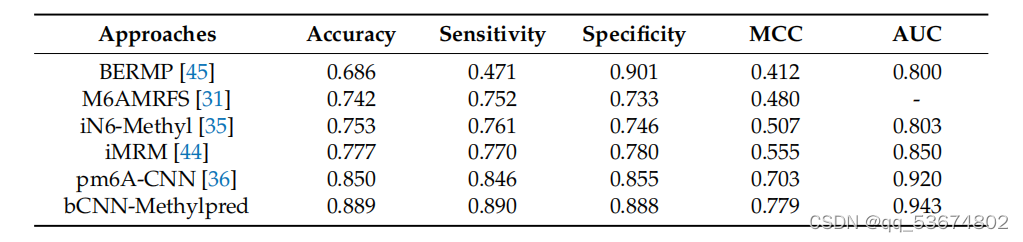
利用基于拟南芥物种的现有方法和现有方法进行m6a位点预测的比较。
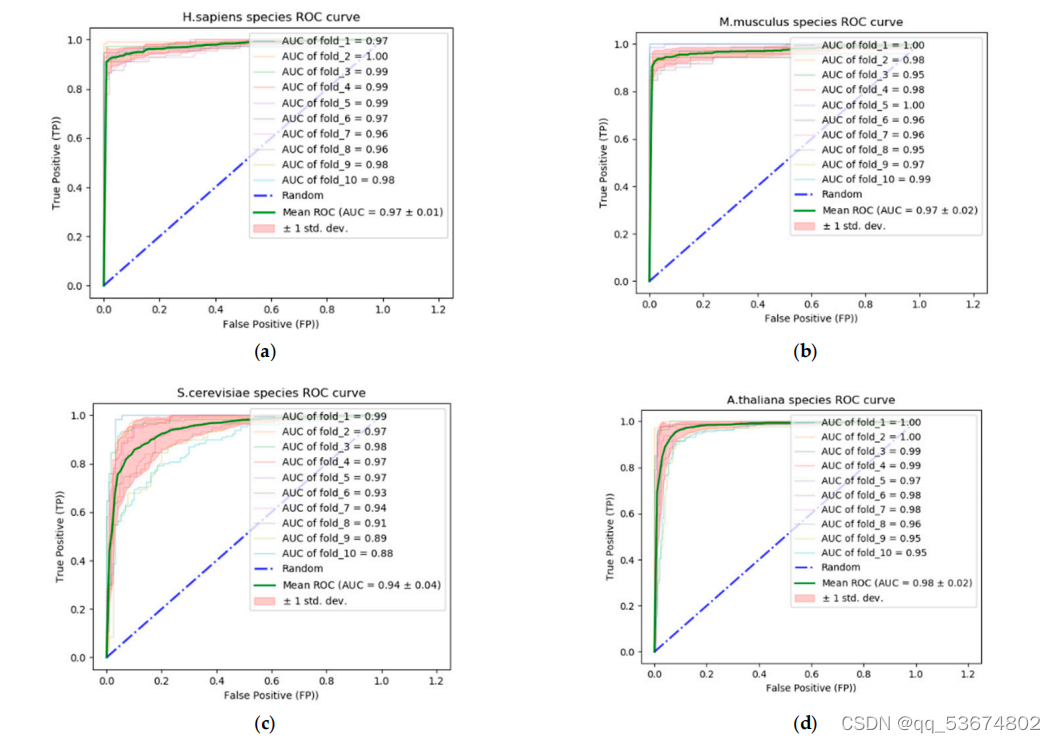
3.4.3.对miCLIP-Seq数据集的预测
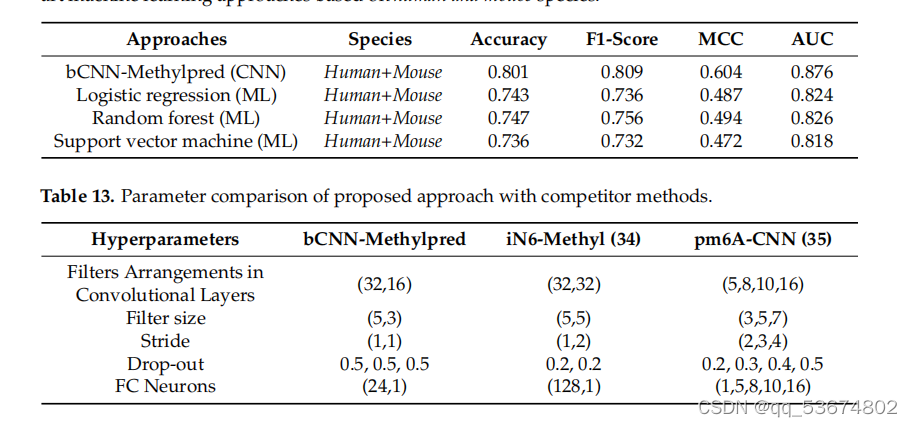
除了上述基准数据集,即智人、肌肉分枝杆菌、酿酒酵母和拟南芥,我们在一个包含人类和小鼠miCLIP-seq数据集的哺乳动物数据集上评估了我们的模型。我们将我们的模型与DeepM6ASeq[43]和其他分类器进行了比较,包括随机森林、逻辑回归和支持向量机,这些都在[43]中提到了。在准确性、f1评分、MCC、AUC方面的总体比较分析列于表11。表11中列出的方法使用了人类和小鼠物种的组合测试样本进行测试,而我们在组合物种和单个物种上测试了我们的模型。基于此分析,我们观察到所提出的模型在准确性、f1分数、MCC和AUC方面都优于DeepM6ASeq[43]和[43]中提到的其他分类器。
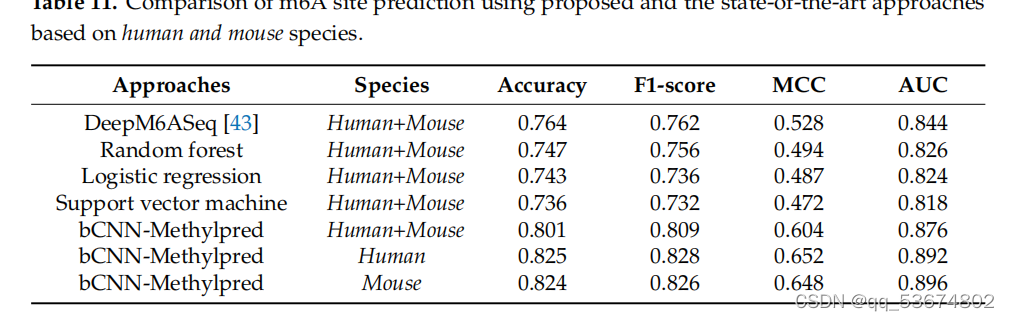
.比较m6a使用基于cnn的方法和基于人类和小鼠物种的最先进的机器学习方法进行站点预测。